Dose-Response Visualiser
Agonist
Observe the effects of changing the characteristics of the Agonist (affinity, intrinsic efficacy) and/or the Cell (receptor density, efficiency of coupling) by moving the red slider along the grey bar to the right or left to increase or decrease the value of the characteristic, respectively.
Some clinically-used drugs act as agonists – e.g. salbutamol at the β2-adrenoceptor, adrenaline at various adrenoceptors, morphine at opioid receptors, etc.
Competitive Antagonist
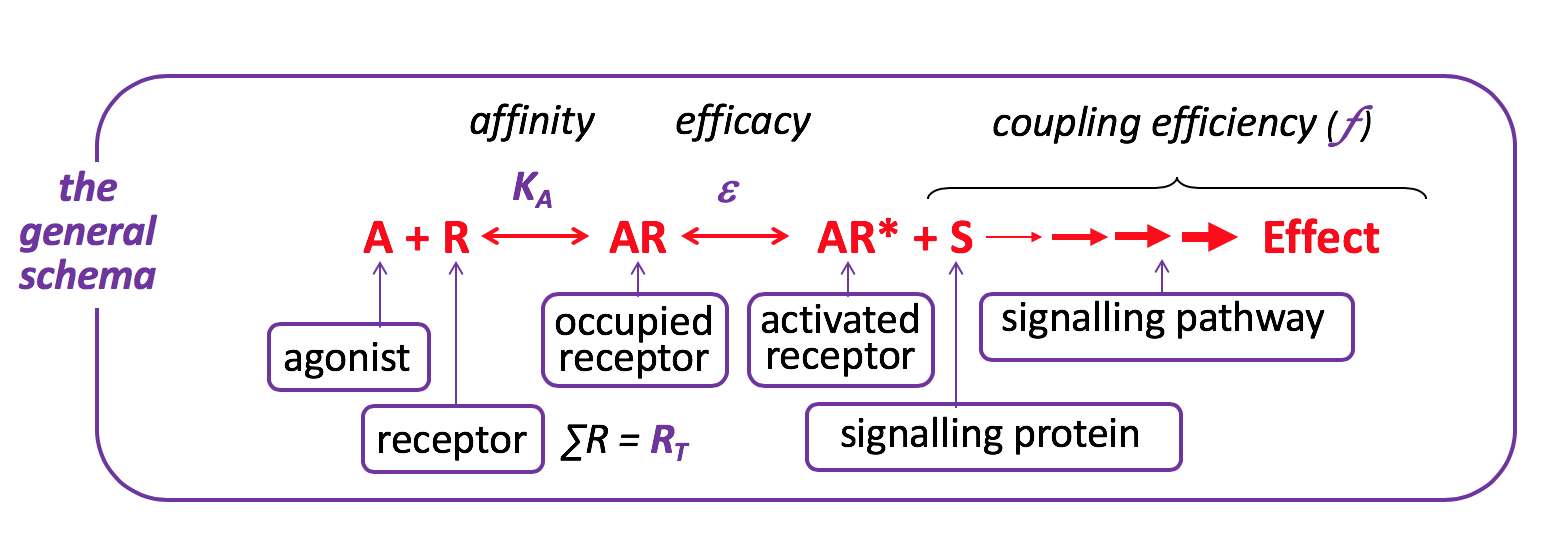
Competitive antagonists bind reversibly to the same binding site on the receptor as the agonist (orthosteric binding). Thus, which molecule occupies the common binding site depends solely on the relative affinity and concentrations of the agonist and competitive antagonist. Thus, theoretically the [agonist] can be raised to the point that the concomitant receptor binding by the antagonist is minimal – at this point the maximum effect of the agonist will be observed, that is competitive antagonism is surmountable. The affinity of a competitive antagonist for a receptor can be determined from binding assays (saturation (KA) and/or competition binding (Ki) assays using radioligands) and from functional assays (KB, by measuring the rightward shift of agonist dose-response curves in the presence of increasing [competitive antagonist] – Schild analysis). Although the constants KA, Ki and KB are determined using different experimental approaches, they each represent the affinity of the competitive antagonist for a receptor, and thus should have the same molar value (the concentration required to occupy 50% of receptors).
The vast majority of clinically-used receptor antagonists are considered to act competitively – e.g. metoprolol at the β-adrenoceptor, atropine at the muscarinic cholinoceptor, losartan at the angiotensin AT1 receptor, etc.
Irreversible Antagonist
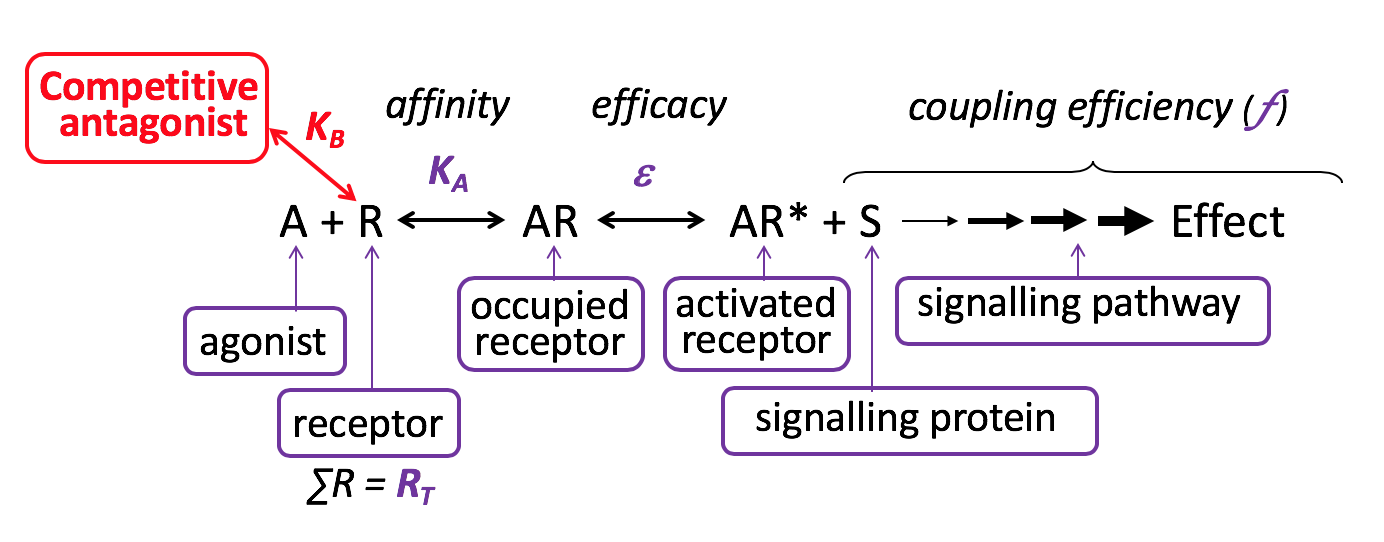
An irreversible antagonist binds covalently (not reversibly) to the same binding site on the receptor as the agonist (orthosteric binding), without activating the receptor. Such antagonists irreversibly inhibit agonist-induced responses, until new functional receptors are expressed by the cell. Irreversible antagonism is one form of non-competitive antagonism.
Relatively few clinically-used receptor antagonists are considered to act irreversibly – e.g. naloxazone at the opioid mu receptor, phenoxybenzamine at the α adrenoceptor. Some drugs act ‘pseudoirreversibly’ and occupy the orthosteric binding site for relatively long periods without forming a covalent bond e.g. tiotropium at the muscarinic cholinoceptor
Allosteric Antagonist (Affinity)
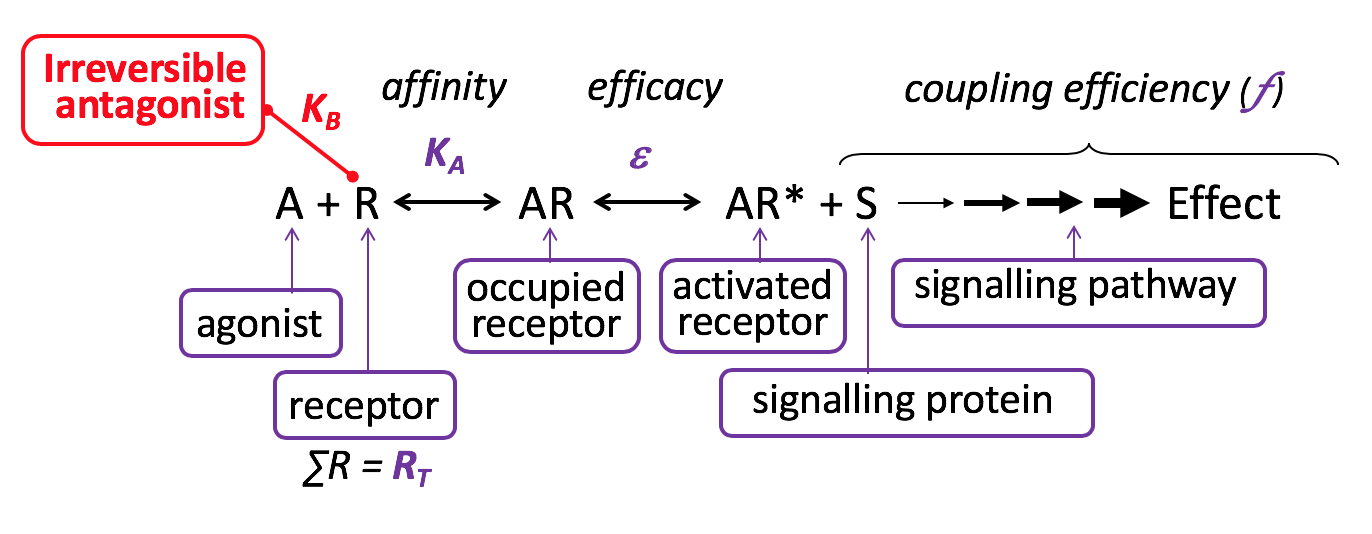
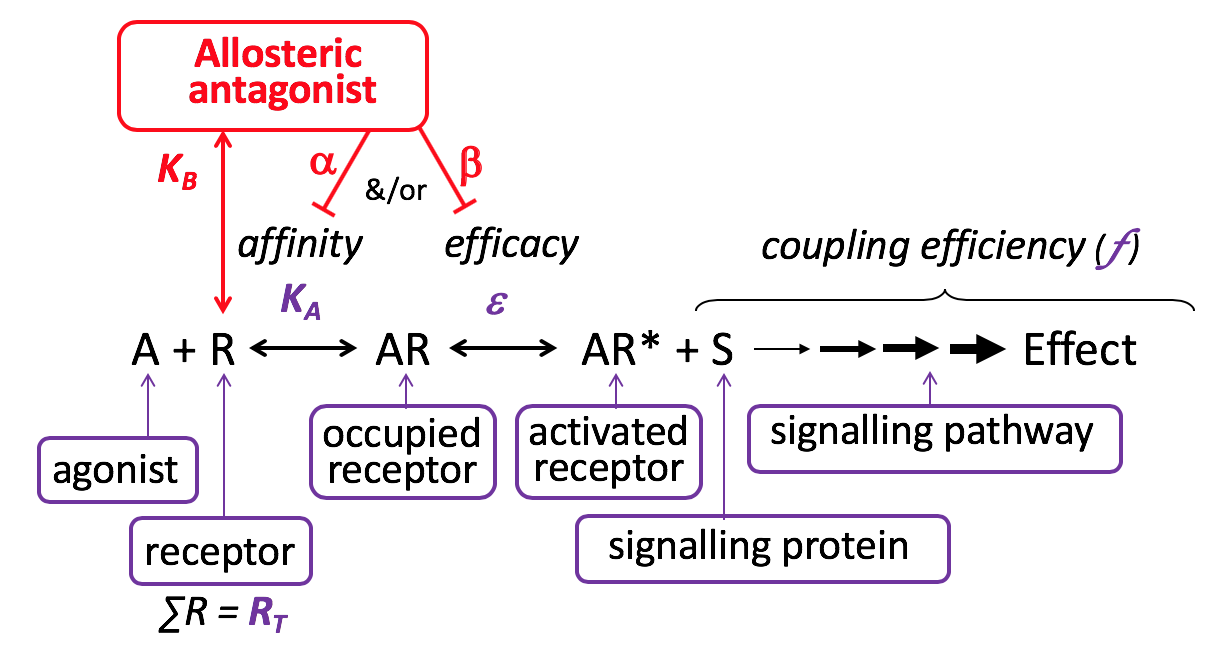
Allosteric antagonists bind reversibly to receptors, without activating the receptor, at a site that is different from the agonist binding site. Allosteric antagonists reduce agonist affinity &/or efficacy, thereby inhibiting agonist-induced effects. They are also referred to as negative allosteric modulators (NAMs). Unlike competitive antagonists, the effects of an allosteric antagonist are saturable – this is because once the allosteric site is saturated, there are no further allosteric effects on the agonist. Allosteric antagonism is another form of non-competitive antagonism, as a receptor can bind both the agonist and the allosteric antagonist simultaneously.
Relatively few clinically-used receptor antagonists are considered to act as negative allosteric modulators although this is an active area of research, e.g. maraviroc at the CCR5 receptor is an allosteric antagonist. In contrast, drugs such as the benzodiazepines act as positive allosteric modulators and enhance the binding affinity of the agonist
Allosteric Antagonist (Efficacy)
Allosteric antagonists bind reversibly to receptors, without activating the receptor, at a site that is different from the agonist binding site. Allosteric antagonists reduce agonist affinity &/or efficacy, thereby inhibiting agonist-induced effects. They are also referred to as negative allosteric modulators (NAMs). Unlike competitive antagonists, the effects of an allosteric antagonist are saturable – this is because once the allosteric site is saturated, there are no further allosteric effects on the agonist. Allosteric antagonism is another form of non-competitive antagonism, as a receptor can bind both the agonist and the allosteric antagonist simultaneously.
Functional Antagonist
Functional antagonists are agonists that produce a functional effect in the cell that is the opposite of that produced by another agonist (e.g. agonist A constricts smooth muscle, whereas agonist B relaxes smooth muscle). The net effect produced depends upon the relative properties of the respective agonists (affinity & efficacy) and their signalling pathways (RT & coupling efficiency).
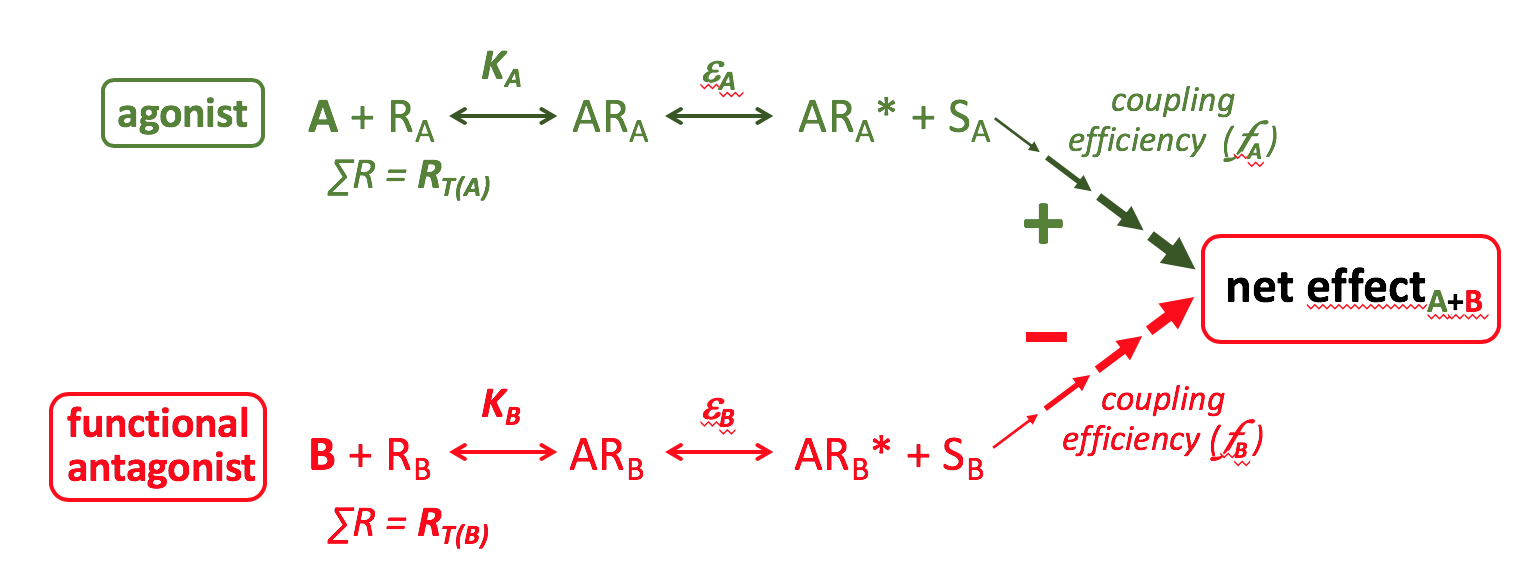
An example of a functional antagonist would be salbutamol, which by activating β2-adrenoceptors on airway smooth muscle and producing relaxation can functionally antagonise airway smooth muscle contraction induced by a wide array of spasmogenic agonists including acetylcholine, histamine and leukotrienes, each acting through their own specific receptors.